基于多模态的3D生成,
寻找骨架新颖、结构合理、与口袋契合的分子;
整合尘封数据及认知,
"动态" 的不断优化待选分子,寻求更高效的人机交互;
MolVadoTM多模态 AI 3D 分子生成模型
基于空口袋的3D分子生成
基于单/多固定片段的3D分子生成
• 符合不同分子设计场景需求:可基于蛋白质空口袋生成结构新颖,构象合理的药物分子;或基于固定的单/多分子片段生成类药分子;
• 遵循工业可用的评估标准:保证模型可基于蛋白质口袋生成与已知活性分子结构相似的化合物,确保生成分子的类药性;
• 生成结果经合理性评估:对生成结果进行2D结构合理性和3D构象合理性评估,过滤结构不合理的分子;
• 支持在线改造&循环生成:可就生成分子进行改造&标记,基于选中分子循环生成,逐步优化目标分子。
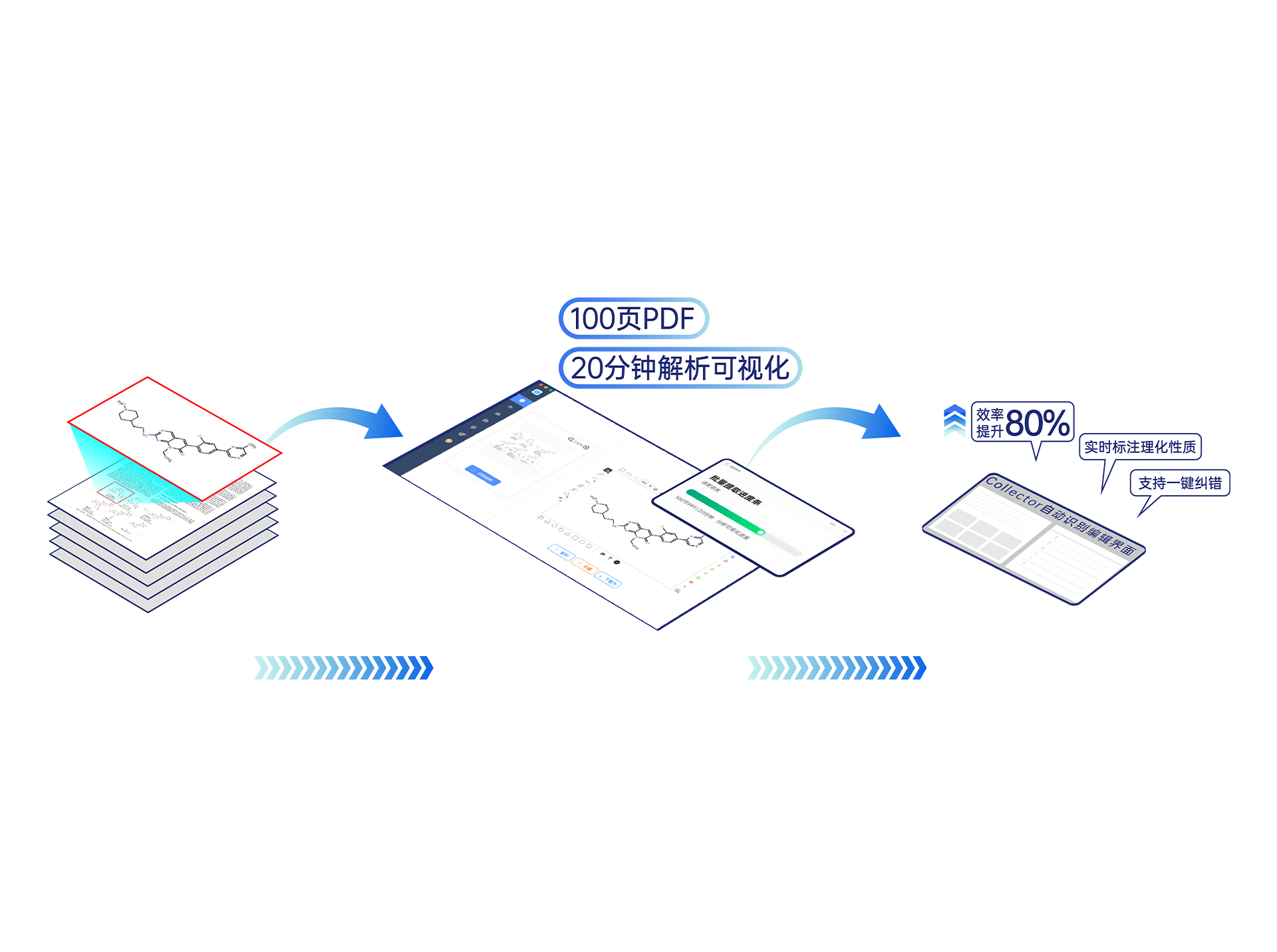
CollectorTM化合物结构&分子信息提取工具
• 桌面级小工具,即开即用:无需联网,随时随地轻松提取结构,识别时间仅需1秒钟左右;
• 高精度分子识别:支持 PDF/图片等多种格式,自动校正模糊图像、低质量扫描件中的结构信息;
• 灵活识别,便捷导出:同时支持OCSR(化学结构)和 IUPAC(标准化学名称)识别,支持SMILES /SDF 多种数据格式快速一键复制。
![]() Win客户端下载
Win客户端下载
![]() Mac客户端下载
Mac客户端下载
![]() 进入网页版
进入网页版
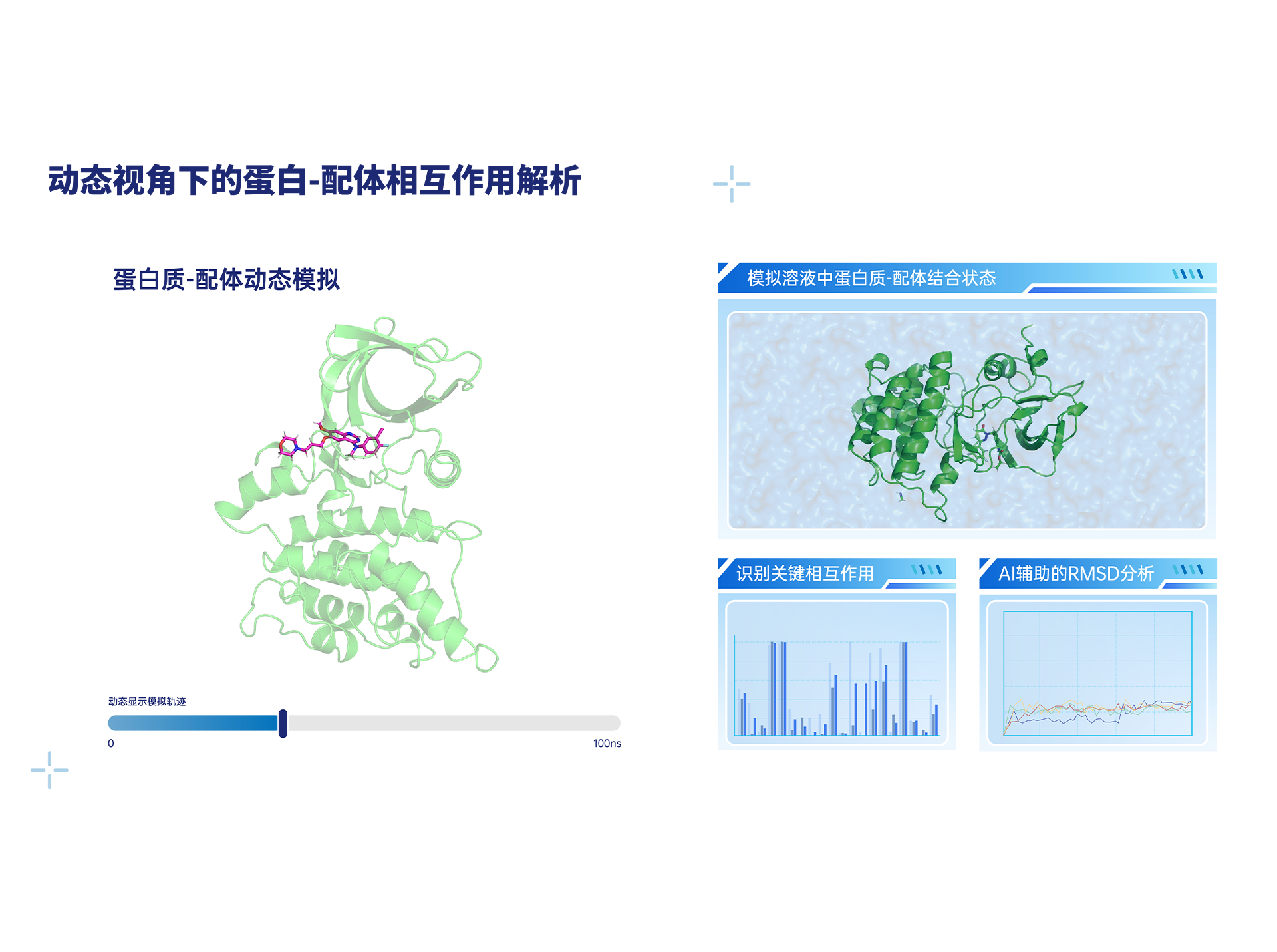
基于分子动力学模拟的蛋白配体相互作用分析
• 动态模拟蛋白质-配体结合状态:突破静态结构局限,通过分子动力学模拟蛋白质-配体结合状态,揭示关键构象变化;
• 非键相互作用识别:解析氢键、π相互作用、盐桥,卤键等非键相互作用,定量评估关键残基对配体亲和力的贡献,为结构优化提供可靠依据;
• AI 辅助高效分析:结合机器学习,从大规模 MD 轨迹中提取关键特征,自动识别最佳结合构象;
• RMSD分析:评估配体自身构象稳定性及其与蛋白质口袋的结合稳定性。
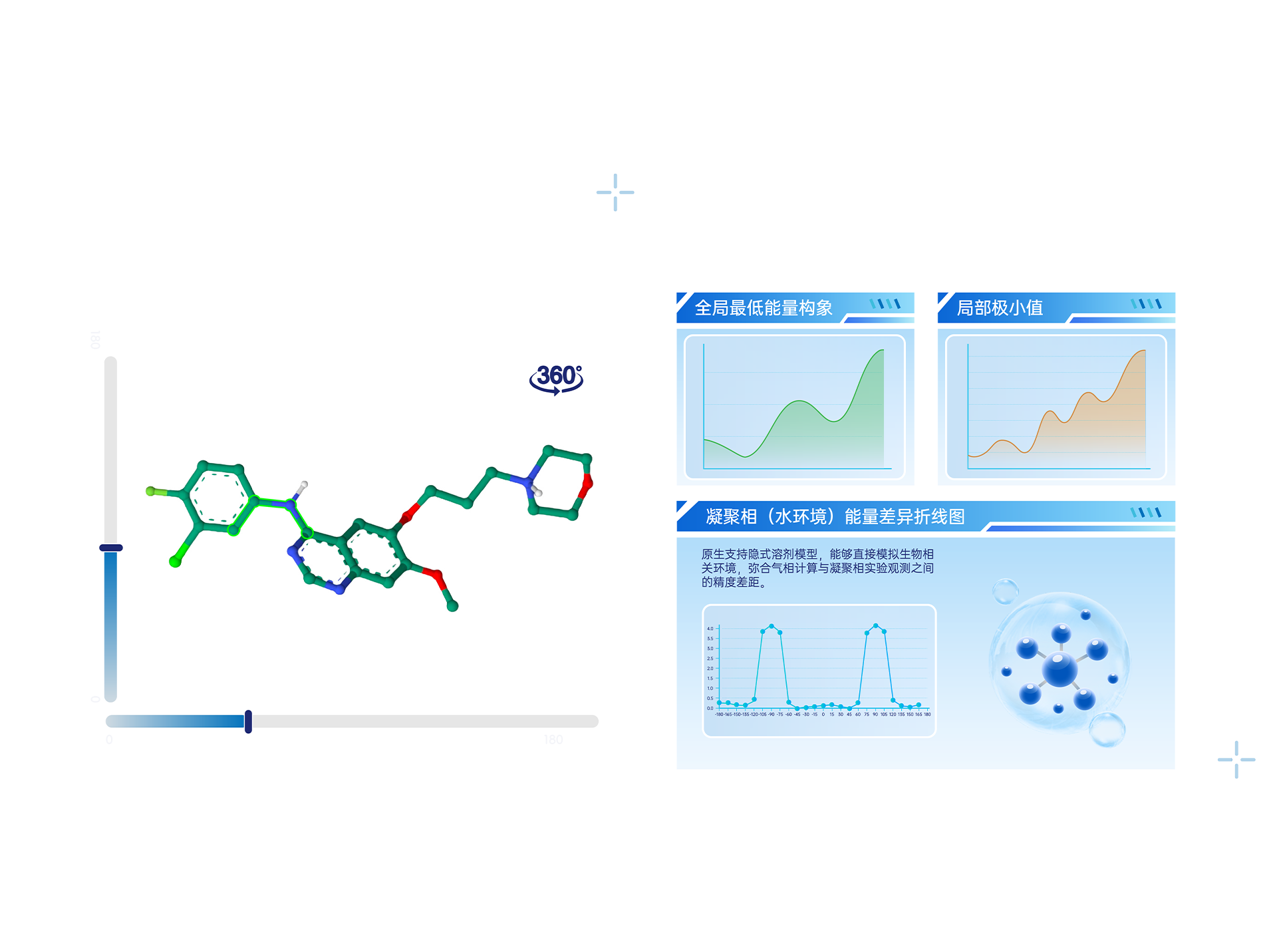
扭转能扫描
• 独创分子切片算法:精准拆分化合物结构,最大限度降低结构误差对扭转能扫描结果的影响;
• 领先的量子化学精度方法:采用双杂化密度泛函理论(DFT)方法,提供接近化学精度的扭转能扫描;
• 无偏差的构象空间探索:通过专有的异步扫描算法消除传统单起点策略的带来的系统性偏差,并结合多副本分子动力学(MD)模拟,准确识别全局能量最低构象;
• 模拟真实生物环境:原生支持隐式溶剂模型,能够直接模拟生物相关环境,弥合气相计算与凝聚相实验观测之间的精度差距。
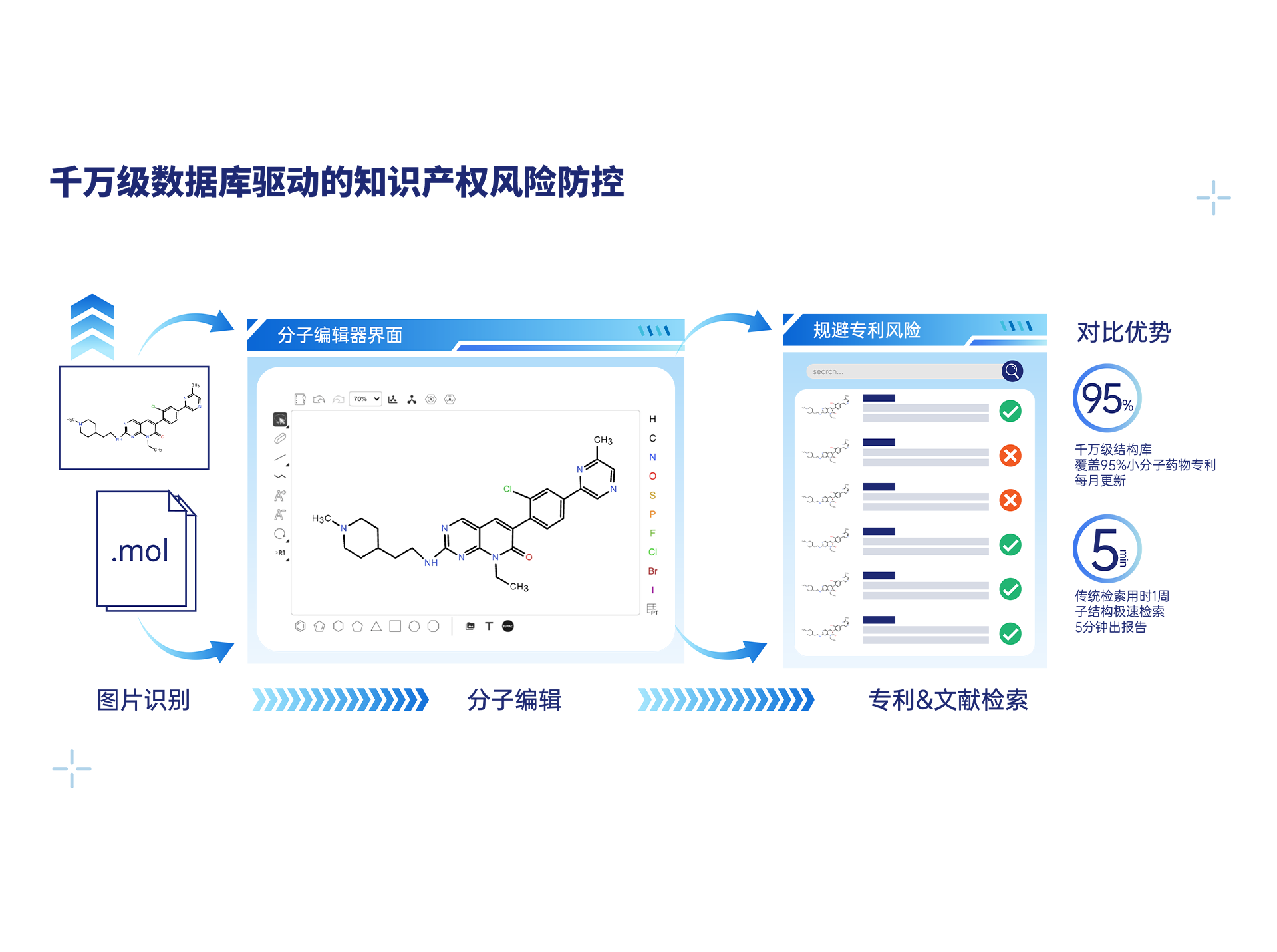
子结构检索
• 超千万级数据库:系统提取与标准化处理专利和文献中的化合物结构,构建超千万级小分子药物结构数据库;
• 专利信息覆盖超95%:数据覆盖95%以上小分子药物专利结构信息,且每月持续更新;
• 支持生成结果及上传结构检索:可基于3D分子生成结果框选分子片段,一站式检索对应的专利和文献信息;
• 全面覆盖专利及文献关键信息:检索结果包含专利公开号,申请日期,公开日期,当前受让人,文献DOI号等。